
Structure - PowerPoint PPT Presentation
1 / 22
Title:
Structure
Description:
A helix forms quickly (10-5 to 10-7 sec) but can unravel almost as quickly. Interestingly formation is generally independent of length, though unraveling isn't. ... – PowerPoint PPT presentation
Number of Views:41
Avg rating:3.0/5.0
Title: Structure
1
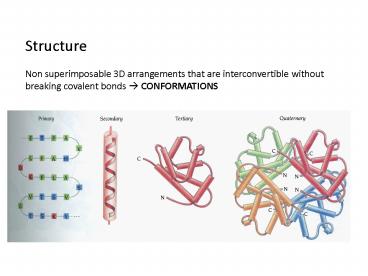
Structure Non superimposable 3D arrangements
that are interconvertible without breaking
covalent bonds ? CONFORMATIONS
2
20 amino acids differing in SIDE CHAINS- these
side chains must confer 3D structure (otherwise
all would look the same!) All amino acids except
glycine are chiral - they can exist in mirror
image forms All backbones are the same L or
D TheL-form reads CORN in clockwise
direction The translational machinery for
protein synthesis has evolved only to
use L-forms
3
Angles and rotations between adjacent
residues N aC f a C C ? C N
? Angle of rotation the only degrees of
freedom are around the aC atoms/bonds. Each
amino acid residue is associated with 2
conformational angles, f and ? Structurally we
like to define f and ? very accurately F and ?
are called dihedral or torsion angles.
4
A polypeptide chain consists of multiple peptide
units, represented by gray boxes. Each subunit
is rigid and planar. The peptide units can
rotate around the Ca along the ? and f angles.
5
So can f and ? have any value? NO!!! Most
combinations of ? and f for an amino acid are not
allowed because of steric collisions between the
side chains and main chain. Theoretically if the
torsion angles all are 180o we have a purely
trans system ( N, Ca, C) if the torsion angles
are all 0 we have the purely cis-
arrangement. Which is preferred? For the
peptide bond (?) Ci Ni1 The
trans is preferred 10001
6
So- proteins like adjacent amino acid side chains
to point away from each other. The exception is
when the i1 residue is a Pro Trans only favored
41 so you can get cis-trans isomerization The
values of f and ? that are possible through the
geometric constraints were first determined by
Ramachandran and are usually plotted against f
and ? angles.
7
Ramachandran Plots Way to visualize dihedral
angles f against ? of amino acid residues in
protein structure. It shows the possible
conformations of f and ? angles for a
polypeptide.
180o
?
0o
-180o
180o
0o
f
8
Certain side chain conformations are
energetically favorable Ethane
Valine Which valine stagger is
energetically the best? The 1st one Why?
9
Following translation we have a primary amino
acid sequence and its random-coil
-- just flopping around folds to a 3D
structure Uses-- VDW interactions
Hydrophobic patches stickiness Salt Bridges
H-bonds Electrostatics
10
Regular conformations of polypeptides MOTIFS OF
PROTEIN STRUCTURE The main driving force for
folding water soluble globular protein molecules
is to pack hydrophobic side chains into the
interior of the molecule thus creating a
HYDROPHOBIC core and a HYDROPHILIC
surface. The problem with creating such a
hydrophobic core from a protein chain is to
bring the side chains into the core the main
chain must also come into the interior. The main
chain is highly polar and therefore hydrophilic,
with one hydrogen bond donor NH and one acceptor
C O for each peptide unit. In a hydrophobic
environment these main chain polar groups must be
neutralized by the formation of hydrogen bonds.
This problem is solved very elegantly by the
formation of regular secondary structure within
the interior.
11
Two Types of 2o structure a helices or ß
sheets Both are characterized by
hydrogen bonding between main chain NH and C O
groups and they are formed when a number of
consecutive residues have the same f and ? angles.
12
a-Helix The right handed a-helix has 3.6
residues per turn and a translation per residue
of 1.5 A which means 5.41A per turn. The atoms
of the backbone pack closely making favorable Van
der Waals interactions.
13
The torsion angles are f -60o and ? -50o
corresponding to the allowed region in the bottom
left quadrant of the Ramachandran plot. These
are hydrogen bonds between C O of residue i and
the NH of residue i 4. Thus all NH and CO
groups are joined with H-bonds except the 1st NH
and last CO group. As a consequence the ends of
the a -helices are polar and are almost always at
the surface of a protein.
Real- slightly bent because not exactly 4
residues/turn but 3.6
Idealized orientation
14
The i to i 4 bonds are 2.9 A long. From the
O? N and are very nearly straight and are nearly
parallel to the helix axis in the classical
helix. Local environments of course can mess
with a-helices somewhat.
15
In natural proteins a slightly different geometry
is seen. The CO groups tend to point out away
from the helix axis and the H-bonds are
consequently not as straight and so f -62o and
? -41o instead of the classical -57 to 60o and
-47 to 50o This geometry actually appears more
stable than the classical a-helix case because it
permits each CO oxygen to H-bond to the NH of the
i 4 residue and also the solvent and/or other
donors. Variations on the classical
a-helix when the chain is more loosely or more
tightly coiled, with hydrogen bonds to residues
i 5 (p-helix) or i 3 (310
helix),respectively, NOT i 4
16
The 310-helix is so called as it has 3 residues
per turn and contains 10 atoms between the
hydrogen bond donor and acceptor. Both the p and
310 helices are rare and usually occur at the
ends of regular helices or as a single turn
helices. Theyre not energetically favorable for
the most partthe backbone atoms are packed too
tight in the 310 or too loose in the p helix that
there is a hole in the middle. a-helices vary in
length from 4/5 residues to over 40
residues. Average 10 ( 3 turns) The rise per
residue of an a-helix is 1.5 A along the helical
axis, which corresponds to about 15 A for an
average helix.
17
An a-helix can in theory be right or left handed
depending on the screw direction of the
chain. Left handed ones dont exist!!!
Because of steric interactions between side
chains and CO groups ALWAYS right handed! (
3-5 residue seen rarely) The a-helix has a
dipole moment All the hydrogen bonds in an
a-helix point in the same direction because the
peptide units are aligned in the same orientation
along the helical axis. Since a peptide unit has
a dipole moment arising from the different
polarity of NH and CO groups, these dipole
moments are also aligned along the helical axis.
The overall effect is a significant net dipole
for the a-helix giving a partial net positive
charge at the amino end and a negative charge at
C-terminal end.
18
The a-helix has a macrodipole moment of n 3.5
Debye units n residues Overall this
translates into a 0.5-0.7 unit charge at each end
of the helix. Expect to attract different
polarity ligands at each end. But ligand
barely binds to C-terminus??
19
Some amino acids are preferred in
a-helices Amino acid side chains project out
from the a-helix and dont interfere with
itexcept for proline. The last atom of the
proline side chain is bonded to the main chain N
atom which forms a ring. This prevents the N
from contributing to a hydrogen bond an adding
steric hindrance to the a-helix conformation.
Proline fits very well into the first (N-term) of
an a-helix and sometimes can be accommodated in
long a-helices by distorting the local geometry
causing a BEND. Pros are often referred to as
helix breakers. Different side chains have a
weak but definite preference either for or
against being in an a-helix. Good helix
formers Ala, Glu, Leu, Met Poor helix
formers Ser Gly -small, flexible-doesnt like
to be in fixed position Try -big and bulky Pro
- cant enter the i ? i 4 groups because cant
H-bond
20
Although they appear structurally to be very
ordered, isolated a-helices are usually
marginally stable in aqueous soltn. A helix
forms quickly (10-5 to 10-7 sec) but can unravel
almost as quickly. Interestingly formation is
generally independent of length, though
unraveling isnt. The most common location
for a helix in a protein is along the outside
with one face out into soltn and one into the
hydrophobic core. -helix wants to have a
hydrophobic and hydrophilic side With the 3.6
residues per turn, there is a tendency for side
chains to change from hydrophobic to hydrophilic
with a periodicity of 3-4 residues. Not a
general hard and fast rule as some helices are
buried but not a bad rule of thumb.
21
With this in mind many a-helices are amphipathic
in that they have predominantly non-polar side
chains along one side of the helical cylinder and
polar residues along the remainder of its
surface. Such helices often aggregate with each
other or with other non-polar surfaces.
Helical wheel- helps to visualize
The view is down the helix with the hydrophobic
core being in the middle. The helix repeats
itself after 5 turns or 18 residues so the 19-21
residues are offset.
22
½ hydrophilic ½ hydrophobic
All polar
Polar charges tend to cluster as do hydrophobics
a-helices that cross membranes are all
hydrophobic. So if we see a sequence of long
stretches of hydrophobics- we can often guess
that these maybe membrane spanning helices.
Recommended
CrystalGraphics Presentations





























